Comparing two genomes in a dotplot
Decomposing genomes in separate blocks provides a very good starting point for pairwise comparison between genomes. The pangenome graph can easily be used to draw a dotplot between two different paths, in which lines represent shared blocks.
In this example we consider the klebs_pangraph.json graph generated from 9 complete chromosomes of Klebsiella Pneumoniae in a previous tutorial.
PyPangraph provides a convenient dotplot function to generate such a dotplot:
import pypangraph as pp
import matplotlib.pyplot as plt
# load pangraph
pan = pp.Pangraph.from_json("klebs_pangraph.json")
# paths to be compared
str_i, str_j = ["NZ_CP013711", "NC_017540"]
fig, ax = plt.subplots(figsize=(8, 8))
pp.dotplot(ax, str_i, str_j, pan, duplicated_color="silver", min_length=150)
plt.show()
Click here to see the full code, in case you want to customize it
import pypangraph as pp
import matplotlib.pyplot as plt
from collections import defaultdict
from itertools import product
from dataclasses import dataclass
@dataclass
class Segment:
"""
Representation for a node of a path as a segment,
with a start and end coordinate and a strandedness.
"""
start: int
end: int
strand: bool
def flip(self):
"""
Flip the segment orientation.
Exchange start and end coordinates and flip the strandedness.
"""
return Segment(self.end, self.start, not self.strand)
def length(self, L):
"""
Calculate the length of the segment.
If total path length L is provided, calculate the length modulo L.
"""
if L is None:
return self.end - self.start
else:
return (self.end - self.start) % L
def crosses_origin(self):
"""
Check if the segment crosses the origin, i.e. start > end
"""
return self.start > self.end
def split_across_origin(self, L):
"""
Split the segment into two segments if it crosses the origin.
"""
assert self.crosses_origin()
s1 = Segment(self.start, L, self.strand)
s2 = Segment(0, self.end, self.strand)
f = s1.length(L) / self.length(L)
return s1, s2, f
def split_fraction(self, f, flip, L):
"""
Split the segment into two segments based on a fraction of the segment length
"""
assert 0 <= f <= 1
l = self.length(L)
l1, l2 = f * l, (1 - f) * l
if flip:
l1, l2 = l2, l1
s1 = Segment(self.start, (self.start + l1) % L, self.strand)
s2 = Segment((self.end - l2) % L, self.end, self.strand)
return s1, s2
def block_segment_dictionary(graph, path_name):
"""
Create a dictionary of block -> segments for a given path in the graph.
"""
path_id = graph.paths[path_name].id
block_sgm = defaultdict(list)
for bid, block in graph.blocks.items():
for nid in block.alignment.node_ids():
node = graph.nodes[nid]
if node.path_id == path_id:
sgm = Segment(node.start, node.end, node.strand)
block_sgm[bid].append(sgm)
return dict(block_sgm)
def linear_plot(ax, sgm_i, sgm_j, **kwargs):
"""
Dotplot for two segments
"""
if sgm_i.strand != sgm_j.strand:
linear_plot(ax, sgm_i, sgm_j.flip(), **kwargs)
else:
ax.plot([sgm_i.start, sgm_i.end], [sgm_j.start, sgm_j.end], **kwargs)
def circular_plot(ax, sgm_i, sgm_j, Li, Lj, **kwargs):
"""
Dotplot for two segments, considering circular genomes.
"""
if sgm_i.crosses_origin():
si1, si2, f = sgm_i.split_across_origin(Li)
flip = sgm_i.strand != sgm_j.strand
sj1, sj2 = sgm_j.split_fraction(f, flip, Lj)
circular_plot(ax, si1, sj1, Li, Lj, **kwargs)
circular_plot(ax, si2, sj2, Li, Lj, **kwargs)
elif sgm_j.crosses_origin():
sj1, sj2, f = sgm_j.split_across_origin(Lj)
flip = sgm_i.strand != sgm_j.strand
si1, si2 = sgm_i.split_fraction(f, flip, Li)
circular_plot(ax, si1, sj1, Li, Lj, **kwargs)
circular_plot(ax, si2, sj2, Li, Lj, **kwargs)
else:
linear_plot(ax, sgm_i, sgm_j, **kwargs)
def dotplot(
ax,
strain_i,
strain_j,
graph,
block_color="black",
no_duplicates=False,
duplicated_color="silver",
min_length=None,
circular=True,
):
"""
Creates a dotplot comparing two paths.
Parameters:
ax (matplotlib.axes.Axes): The matplotlib axes object where the plot will be drawn.
strain_i (str): The identifier for the first path.
strain_j (str): The identifier for the second path.
graph (Graph): pangenome graph object.
block_color (str, optional): The color used for non-duplicated blocks. Defaults to "black".
no_duplicates (bool, optional): If True, duplicated blocks will not be plotted. Defaults to False.
duplicated_color (str, optional): The color used for duplicated blocks. Defaults to "silver".
min_length (int, optional): Minimum length of blocks to be plotted. Defaults to None.
circular (bool, optional): If True, plots the segments in a circular layout. If False, uses a linear layout. Defaults to True.
"""
# Create block segment dictionaries for the paths
bs_i = block_segment_dictionary(graph, strain_i)
bs_j = block_segment_dictionary(graph, strain_j)
# Get the nucleotide lengths of the paths
Li = graph.paths[strain_i].nuc_len
Lj = graph.paths[strain_j].nuc_len
# Plot the segments
for bid, seg_i in bs_i.items():
if bid not in bs_j:
continue
color = block_color
seg_j = bs_j[bid]
if min_length is not None:
# optionally skip short blocks
block_len = len(graph.blocks[bid].consensus())
if block_len < min_length:
continue
if (len(seg_i) > 1) or (len(seg_j) > 1):
if no_duplicates:
continue
# color duplicated blocks in gray
color = duplicated_color
for si, sj in product(seg_i, seg_j):
if circular:
circular_plot(ax, si, sj, Li, Lj, color=color)
else:
linear_plot(ax, si, sj, color=color)
# Load the Pangraph from a JSON file
pan = pp.Pangraph.from_json("klebs_pangraph.json")
# paths to be compared
str_i, str_j = ["NZ_CP013711", "NC_017540"]
fig, ax = plt.subplots(figsize=(8, 8))
dotplot(ax, str_i, str_j, pan)
plt.tight_layout()
plt.show()
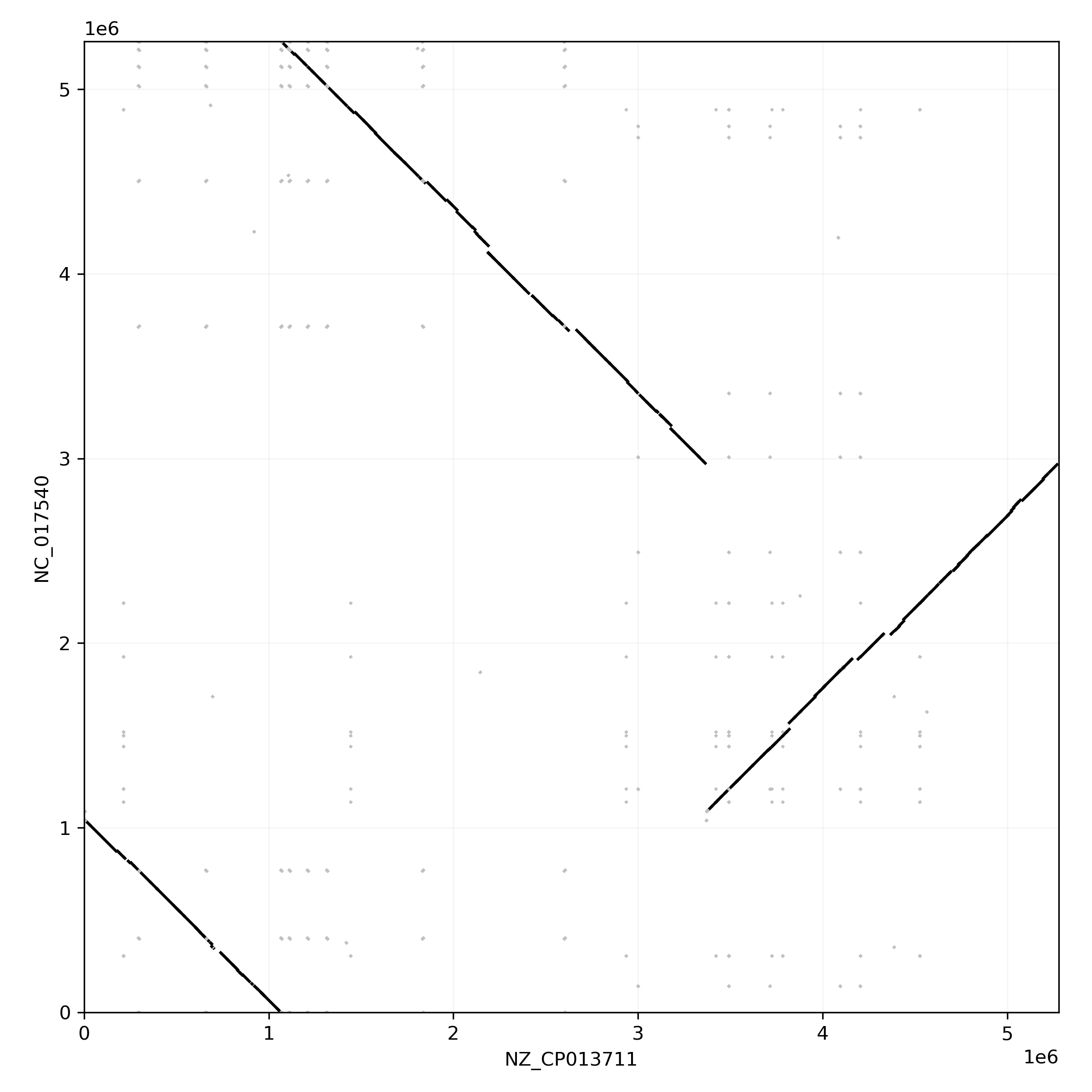
Block that are not duplicated between the two paths are displayed in black. We observe that the chromosomes differ in a large inversion.
Duplicated blocks are depicted in gray. These form a checkerboard pattern, typical of blocks that are repeated multiple times on the genome. We can get the list of most duplicated blocks using the to_blockstats_df method:
df = pan.to_blockstats_df()
df[df["duplicated"]].sort_values("count", ascending=False).head(3)
# block_id count n_strains duplicated core len
# 3121046370622183165 101 9 True False 236
# 17902739066067298048 72 9 True False 1733
# 9722346788777533867 72 9 True False 3062
It is also easy to retrieve the consensus sequence of one of these blocks with:
b = pan.blocks[17902739066067298048]
print(b.consensus())
# TTCATCAGACAATCTGTGTGAGCACTACAAAGGCAGGTTCTTTAAGGTAAGGA...
Nucleotide blast of this sequence reveals a match with the 16S ribosomal RNA gene.