Exporting sequences
Pangraph's export command also allows the export of nucleotide sequences from the graph. We will cover:
- exporting the consensus sequences of all blocks
- exporting all node sequences from all blocks, aligned or not.
- exporting the core-genome alignment and building a core-genome tree.
Exporting block consensus sequences
Block consensus sequences can be exported using the export block-consensus subcommand:
pangraph export block-consensus \
graph.json \
-o block_cons.fa
This generates the block_cons.fa FASTA file. This file contains one entry per block, with the block ID as the header and the consensus sequence as the sequence:
>9245376340613946
ATTTCCGGTGATTAAGTCTGAGGATTTT...
>12899102400345352
ATCAGACCGCTTCTGCGTTCTGATTTAA...
>19466549525654178
AAAGGTTGCTTGCCGAACGATTCGTGGT...
...
This file can be used for example to search for homology with a particular query sequence, e.g. a gene of interest.
Exporting block alignments
In addition to the block consensus sequences, alignments for each block can be exported using the export block-sequences subcommand:
pangraph export block-sequences \
graph.json \
-o block_aln
This generates a block_aln directory containing one FASTA file per block, named block_<block_id>.fa. Each of these files contains the alignment for the block:
>324496867494915323 {"path_name":"NC_013361","block_id":158121245228334622,"start":4113083,"end":4113579,"strand":"+"}
ATTCATGTCCTTGACTGCTTTGTTAATGTCGCACTGGA...
>1447999556612276352 {"path_name":"NZ_CP011342","block_id":158121245228334622,"start":3167087,"end":3167583,"strand":"+"}
ATTCATGTCCTTGACTGCTTTGTTAATGTTGATCTGGA...
>3256239825927255217 {"path_name":"NZ_CP015912","block_id":158121245228334622,"start":2110489,"end":2110985,"strand":"+"}
ATTCATGTCCTTGACTGCTTTGTTAATGTCGCACTGGA...
...

The FASTA id of each entry is the node id, while the description contains a json string with additional information: the path name, block id, start and end positions of the node, and strandedness.
Note that while these alignments contain deletions, they do not include insertions. This is due to the fact that alignments are relative to the block consensus, against which insertions cannot be placed (see the previous tutorial section). However pangraph also provides the option to export complete, but unaligned, sequences for each block:
pangraph export block-sequences \
graph.json \
--unaligned \
-o block_seqs
Similarly to the previous command, this generates a block_seqs directory containing one FASTA file per block. In this case each file contains the complete but unalligned set of sequences for the block, including insertions. These can then be aligned with other tools of your choice, such as MAFFT or ClustalW.
Exporting the core-genome alignment and building a tree
Pangraph also provides a quick command to extract the core-genome alignment of the graph: the export core-genome subcommand. This is defined as the concatenated alignment of all single-copy blocks that are present exactly once in all paths. The order and strandedness in which these blocks are concatenated can be specified using the --guide-strain option. This option takes the name of a path in the graph, and the blocks are ordered according to their order in that path.
pangraph export core-genome \
graph.json \
--guide-strain NC_010468 \
-o core_genome_aln.fa
Also the export core-genome subcommand has the --unaligned optional flag. When activated, the complete (i.e. including insertions) but unaligned core-genome sequences are exported. These can then be aligned by the user with its preferred tool.
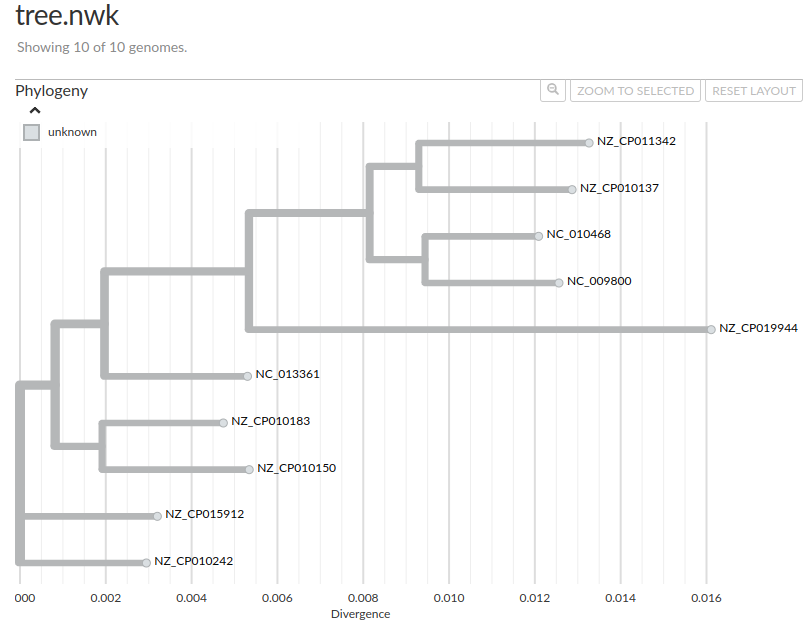
This alignment can be used for example to build a phylogenetic tree for the dataset1. For example using FastTree:
fasttree -nt -gtr core_genome_aln.fa > tree.nwk
Here is the tree, visualized with auspice: